























Pengidap thalasemia harus melakukan transfusi darah dan mengkonsumsi obat seumur hidupnya. Penyakit kelainan darah ini masih sangat susah untuk disembuhkan. Memutus rantai keturunan thalasemia harus dilakukan dengan skrining dan niat untuk melakukan pencegahan.
Kategori
Cek Kesehatan
Komunitas
Postingan Utama
Lihat selanjutnyaBooking Dokter

Tantangan Memutus Rantai Penyakit Thalasemia di Indonesia

Thalasemia tidak bisa disembuhkan, pencegahan jadi prioritas

Pada 1921, dokter spesialis anak dan hematologi, Thomas Benton Cooley, melihat ada keanehan pada beberapa pasien bayi dan anak di Children’s Hospital Michigan, Amerika Serikat. Pasien-pasien anak ini mengalami anemia disertai gejala perut yang membengkak, tulang membesar, dan kulitnya menjadi gelap. Ia kemudian mulai melakukan penelitian tentang bentuk anemia pada empat anak keturunan Italia dan Yunani yang memiliki kesamaan dalam perubahan tulang.
Empat tahun kemudian, ia mempresentasikan temuannya kepada Asosiasi Dokter Anak Amerika dengan menyebut kelainan ini sebagai anemia eritroblastik atau anemia mediteranean atau anemia Cooley.
Hampir seabad setelah penemuan dokter Cooley, penyakit kelainan darah ini masih belum ditemukan obatnya dan dinyatakan tidak bisa disembuhkan.
Anemia Cooley atau thalasemia adalah penyakit kelainan darah yang disebabkan oleh rusak atau cacatnya rantai gen yang menyusun hemoglobin yaitu rantai alfa dan beta.
Ada dua jenis thalasemia yakni thalasemia alfa (α-) dan thalasemia beta- (β-). Thalasemia alfa terjadi ketika ada 1 atau lebih hemoglobin yang rusak, sebab rantai alfa dalam darah memerlukan 4 gen hemoglobin sehat. Sedangkan rantai beta dalam darah memerlukan 2 gen sehat, jika salah satu dari kedua gen tersebut rusak atau bermutasi maka akan terjadi thalasemia beta.
Selain berdasarkan mutasi genetik, thalasemia dapat pula dibagi berdasarkan tingkat keparahan manifestasinya. Penyakit thalasemia minor (ringan) akan terjadi ketika anak mewarisi satu sel globin yang rusak dari salah satu orang tuanya, baik ibu atau bapak.
Sedangkan penyakit thalasemia mayor (berat) akan terjadi ketika anak mewarisi sel globin rusak dari kedua orang tua yang memiliki thalasemia.
Kondisi thalasemia minor bisa saja tidak menimbulkan gejala apapun. Namun sepasang suami istri yang memiliki thalasemia minor dapat menurunkan thalasemia mayor pada anaknya. Pada beberapa kondisi, mutasi pada kedua sel alfa dapat menyebabkan keadaan yang disebut hydrops fetalis. Kondisi ini dapat menyebabkan kematian pada janin.
Penyakit ini masih sangat sulit untuk disembuhkan. Ada beberapa pilihan terapi kuratif yang dapat diberikan (seperti cangkok sumsum tulang), tetapi harganya masih sangat mahal dan belum dapat dilakukan di Indonesia.
Pencegahan hanya bisa dilakukan dengan melakukan skrining untuk mengetahui kondisi dan menghindari pernikahan sesama pembawa sifat thalasemia. Salah satu faktor risiko yang paling banyak dilaporkan dari thalasemia adalah pernikahan kerabat. Anak yang lahir dari perkawinan sedarah diperkirakan menerima salinan gen yang identik dari kedua orang tuanya.
Thalasemia di Indonesia
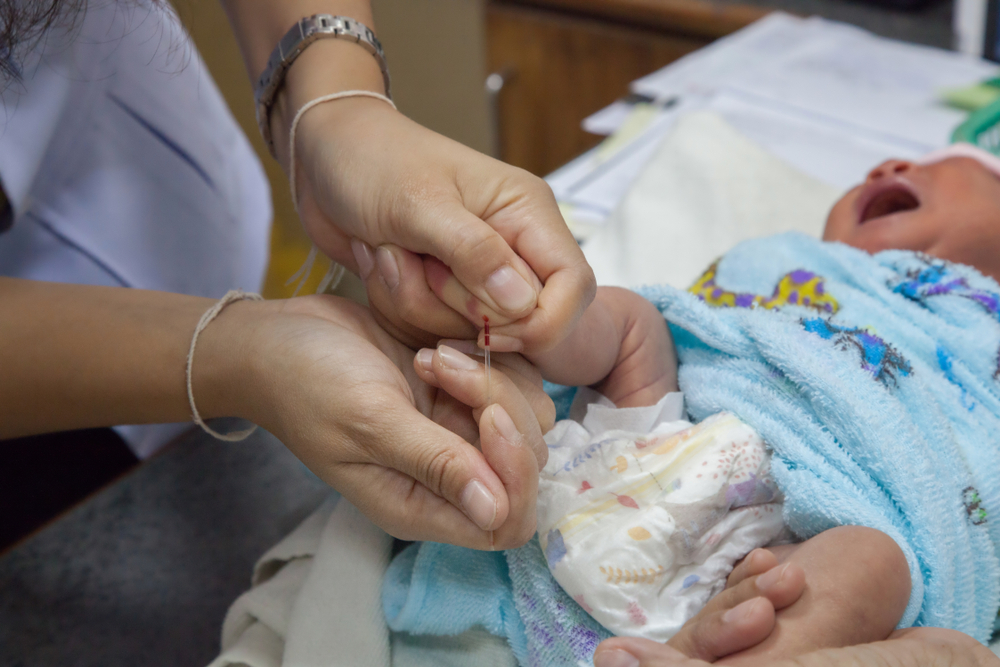
Untuk penyakit yang terbilang cukup langka, Indonesia termasuk negara di thalassemia belt alias wilayah yang memiliki risiko tinggi thalasemia. Setiap tahunnya, jumlah kasus thalasemia di Indonesia terus meningkat.
Berdasarkan penelitian dari Prof. Iskandar Wahidiyat dan Prof. Pustika Amalia Wahidiyat dari Fakultas Kedokteran Universitas Indonesia, diketahui bahwa frekuensi carrier atau pembawa genetika thalasemia mencapai 5% dari seluruh populasi di Indonesia. Pernikahan sesama pembawa sifat thalasemia berisiko mewariskan penyakit thalasemia pada anaknya.
Selain menjadi bahaya seumur hidup pada pengidapnya, penyakit yang tidak bisa disembuhkan ini memakan biaya pengobatan yang sangat tinggi. Beban biaya pengobatan thalasemia menempati nomor 5 terbesar di Indonesia. Kementerian kesehatan mencatat, pembiayaan JKN (Jaminan Kesehatan Nasional) untuk penanganan thalasemia periode 2014-2018 menelan biaya hingga lebih dari Rp 2 triliun.
Pencegahan untuk memutus rantai thalasemia masih menjadi tantangan tersendiri bagi Indonesia. Satu-satunya cara pencegahan yang bisa dilakukan adalah dengan menghindari pernikahan sesama pembawa sifat thalasemia (carrier) dan sesama penyitas thalasemia. Para carrier hanya dapat mengetahui bahwa mereka membawa sifat thalasemia dengan melakukan pemeriksaan skrining thalasemia (pemeriksaan hemoglobinopati).
Tapi berapa banyak orang yang tidak sakit dan dengan kesadaran sendiri secara sukarela melakukan skrining hemoglobinopati?
Tantangan skrining thalasemia
Skrining thalasemia saat ini hanya tersedia di puskesmas-puskesmas yang ada di kota besar dengan biayanya yang relatif mahal, yakni berkisar Rp 400.000 – Rp 500.000. Meski begitu angka tersebut tetap terbilang murah jika dibandingkan dengan risiko tinggi yang akan dialami jika mengidap thalasemia.
WHO memberi rekomendasi kepada negara seperti Indonesia untuk mengadakan program skrining thalasemia rutin. Namun sejauh ini belum ada program nasional yang signifikan dari pemerintah dalam melakukan skrining untuk memutus rantai thalasemia.
Sementara itu negara-negara tetangga seperti Malaysia dan Thailand sudah mewajibkan skrining thalasemia dalam rangkaian tes kesehatan pra-nikah sebagai bagian dari program pemerintah.
Agar program skrining tepat sasaran, para pelaksana harus mengetahui terlebih dahulu persepsi dan pemahaman masyarakat setempat. Oleh karena itu, informasi tentang knowledge, attitude, dan practice (KAP) masyarakat sasaran menjadi sangat penting.
Penelitian oleh Unpad pada 2015 menemukan bahwa 9,3% siswa SMA di Jatinangor dan 8% siswa SMA di Banyumas mengidap thalasemia beta minor. Penelitian pada mahasiswa FKUI menemukan bahwa 5,3% di antaranya merupakan carrier. Belum ditemukan penelitian yang fokus pada kelompok usia bekerja di Indonesia.
Di PubMed, hanya tercatat 1 penelitian (tahun 2011) yang meneliti tentang KAP thalasemia pada masyarakat di pulau Jawa. Dengan kata lain, penelitian tentang persepsi dan kesadaran masyarakat Indonesia untuk memutus thalasemia masih sangat terbatas, padahal prevalensinya tinggi. Dalam konteks kesehatan masyarakat, data primer dari penelitian ini mampu menjadi landasan studi lanjutan maupun program intervensi yang lebih efektif.
Catatan
Hello Sehat tidak menyediakan saran medis, diagnosis, atau perawatan. Selalu konsultasikan dengan ahli kesehatan profesional untuk mendapatkan jawaban dan penanganan masalah kesehatan Anda.
Versi Terbaru
20/07/2022
Ditulis oleh dr. Mikhael Yosia, BMedSci, PGCert, DTM&H.
Diperbarui oleh: Abduraafi Andrian
Ditulis oleh
dr. Mikhael Yosia, BMedSci, PGCert, DTM&H.
General Practitioner · Medicine Sans Frontières (MSF)
 Iklan
IklanApakah artikel ini membantu?
IklanIklan